CASE REPORT | https://doi.org/10.5005/jp-journals-10071-23329 |
Hemophagocytic Lymphohistiocytosis Related to Tuberculosis Disease
1Department of Pediatric Critical Care, University of Health Sciences, Ümraniye Training and Research Hospital, Istanbul, Turkey
2Department of Pediatric Infectious Disease, University of Health Sciences, Ümraniye Training and Research Hospital, Istanbul, Turkey
3,4Department of Pediatrics, University of Health Sciences, Ümraniye Training and Research Hospital, Istanbul, Turkey
5Department of Pediatric Radiology, University of Health Sciences, Ümraniye Training and Research Hospital, Istanbul, Turkey
6Department of Pediatric Hematology and Oncology, University of Health Sciences, Ümraniye Training and Research Hospital, Istanbul, Turkey
7Department of Pediatric Rheumatology, University of Health Sciences, Ümraniye Training and Research Hospital, Istanbul, Turkey
Corresponding Author: Seher Erdoğan, Department of Pediatric Critical Care, University of Health Sciences, Ümraniye Training and Research Hospital, Istanbul, Turkey, Phone: +90 532-6678370, e-mail: seher70@gmail.com
How to cite this article Erdoğan S, Çakır D, Bozkurt T, Karakayalı B, Kalın S, Koç B, et al. Hemophagocytic Lymphohistiocytosis Related to Tuberculosis Disease. Indian J Crit Care Med 2020;24(1):63–65.
Source of support: Nil
Conflict of interest: None
ABSTRACT
Hemophagocytic lymphohistiocytosis (HLH) is a rare, albeit potentially fatal, condition in which fever, hepatosplenomegaly, and cytopenia predominate the clinical picture. Although it may be primary, it may also develop secondary to various etiologies. Herein, we aimed to report a patient who was diagnosed with pulmonary tuberculosis, developed fever and cytopenia during follow-up, and received immunomodulatory therapy together with antituberculosis therapy for the diagnosis of HLH. Sequencing of PRF1 showed heterozygous mutation. Although primary HLH has been detected in infants and children, genetic mutation of genes should be considered a differential diagnosis of HLH even in the adolescent.
Keywords: Hemophagocytic lymphohistiocytosis, Therapeutic plasma exchange, Tuberculosis.
INTRODUCTION
Hemophagocytic lymphohistiocytosis (HLH) is characterized by a clinical picture where fever, hepatosplenomegaly, and cytopenia predominate as a result of an impairment of the function of cytotoxic T-lymphocytes and natural killer (NK) cells, activation of macrophages and T-lymphocytes, excessive production of pro-inflammatory cytokines, and hemophagocytosis.1 It is categorized as primary (familial) and secondary. Secondary HLH may occur due to viral, bacterial, parasitic, fungal, protozoal diseases as well as malignancy, radical stress, metabolic disorders, immune deficiency, and collagen tissue disorders.
Hemophagocytic lymphohistiocytosis is characterized by a systemic cytokine storm [interleukin (IL)-1β, tumor necrosis factor alpha (TNFα), IL-6, IL-8, interferon gamma (IFNγ), IL-18, and IL-10] leading to organ dysfunction. Patients receive the HLH-2004 treatment protocol during this time. In some cases, this protocol is insufficient for the suppression of inflammation and mortality occurs. Suppression of disease activity can be achieved with the removal of cytokines with plasma filtration in such patients.2 Herein, we report an HLH case that developed secondary to tuberculosis infection and received therapeutic plasma exchange (TPE) in an attempt to remind clinicians that HLH must be considered in the differential diagnosis of pancytopenia in severe tuberculosis cases.
CASE DESCRIPTION
An 11-year-old male patient presented to a healthcare facility with fever and cough for 2 days and had been referred to our hospital for a preliminary diagnosis of complicated pneumonia. On physical examination, he weighed 30 kg, body temperature was 39°C, maximum heart rate was 125 bpm, respiratory rate was 30 breaths per minute, and a blood pressure reading of 115/77 mm Hg was noted. On auscultation, he had diminished breath sounds over the right hemithorax. There were palpable submandibular lymph nodes of 1.5 cm × 0.5 cm on both sides and palpable right axillary lymph node of 1 cm × 0.5 cm in size. Other physical examination findings were normal. Complete blood count results were as follows: leukocyte count, 8290/mm3; hemoglobin, 9.8 g/dL; and platelet count, 545000/mm3. The sedimentation rate was 38 mm/hour. A thoracic ultrasonography revealed septated pleural fluid with a dense content, which reached a depth of up to 30 mm. A thoracic catheter was placed, and intrapleural tissue plasminogen activator was administered three times. Intravenous ceftriaxone and clindamycin were administered. A thoracic computed tomography was taken, showing multiple lymph nodes having a conglomerated appearance, the largest of which measured 18 mm × 13 mm, in the mediastinal pretracheal, subcarinal, and right hilar regions. There existed a dense pleural effusion with a depth of up to 35 mm in the basal part of the right hemithorax. We also observed intensive pleural fluid in the right hemithorax from apex to the basale in coronal and sagittal images (Figs 1 and 2). As the pleural biopsy material was necrotic and mycobacteria polymerase chain reaction was positive, pathological examination could not be done. An echocardiogram revealed normal findings. As the patient’s fever and respiratory difficulty persisted and his radiological signs did not improve, his antibiotic therapy was switched to piperacillin plus teicoplanin. Pleural fluid and blood cultures were negative. Serological tests for human immunodeficiency virus, Epstein–Barr virus, cytomegalovirus, hepatitis B and C, chlamydia, and mycoplasma were all negative. His clinical symptom did not improve with nonspecific therapy, and he was started on pyrazinamide, isoniazid, rifampicin, and ethambutol as antituberculosis treatment empirically. He could not use antituberculous therapy for the first week due to vomiting.

Fig. 1: Irregularly pleural thickness and parenchymal indentation
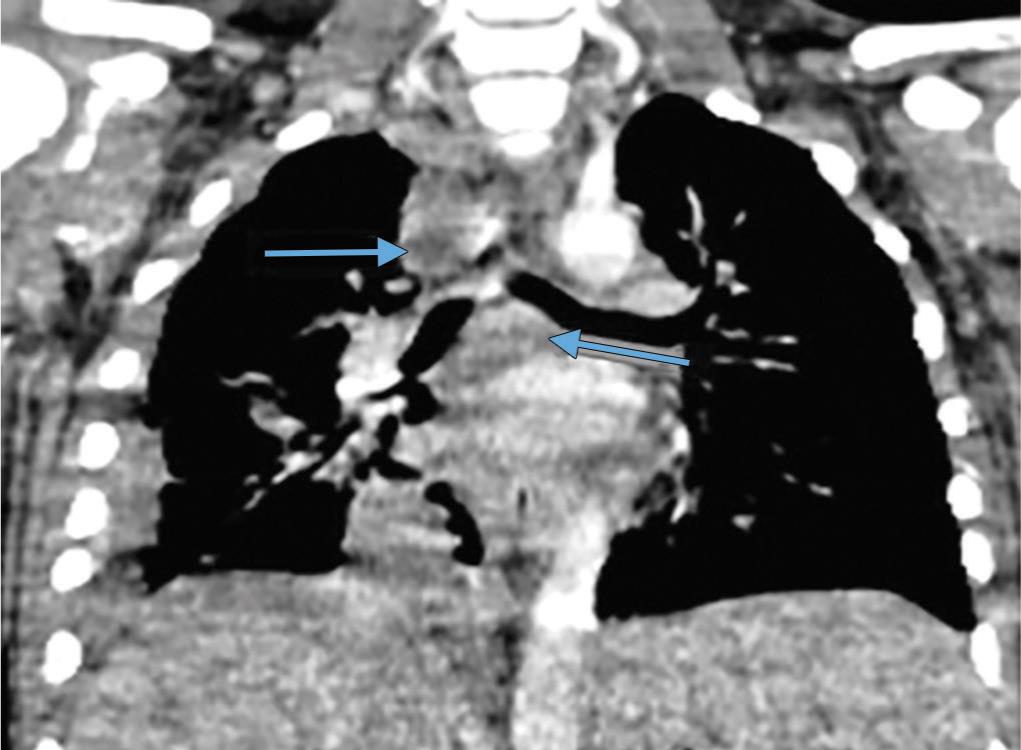
Fig. 2: Coronal and axial images show paratracheal and subcarinal mediastinal grown lymph nodes with central necrosis
Purified protein derivative measured 10 mm; acid-resistant bacilli were negative in the sputum and fasting gastric juice. He had QuantiFERON positive. His family screen for tuberculosis turned negative. He was noted to have bicytopenia in full blood count on the 35th day of follow-up, and a bone marrow aspiration was performed, which revealed signs of dysplasia in normoblasts and two hemophagocytosis. His laboratory results were as follows: ferritin: 11.735 ng/mL (N: 5.6–216), fibrinogen: 159 mg/dL (N: 200–400), alanine aminotransferase (ALT): 154 U/L (N: 0–55), aspartate aminotransferase (AST): 1,196 U/L (N: 5–34), gamma-glutamyl transferase: 464 U/L (38–54), cholesterol: 102 mg/dL, triglyceride: 164 mg/dL, lactic acid dehydrogenase (LDH): 2936 U/L (N: 110–295), albumin: 2.4 g/dL (3.5–5), direct bilirubin (D.Bil): 1.78 mg/dL, c-reactive protein (CRP): 6.6 mg/dL (N: 0–0.5), procalcitonin: 16.4 ng/mL (N: %3C;0.05), sodium: 132 mEq/L (N: 138–145), and international normalized ratio (INR): 1.62. A hemophagocytic syndrome secondary to tuberculosis infection was primarily considered. As his fever did not subside and his tachypnea persisted, he was transferred to the pediatric intensive care unit. There he was started on intravenous immunoglobulin (IVIG) 400 mg/kg/day for 5 days. An 11.5 Fr hemodialysis catheter was placed, and 10 sessions of TPE with adjunctive fresh frozen plasma were performed. A cervical lymph node biopsy was taken, which was consistent with tuberculosis infection. As his clinical condition did not improve, dexamethasone 10 mg/m2 was commenced. Subsequently, during follow-up, his fever subsided, rash, tachypnea eliminated, and appetite returned to normal. Subsequent laboratory values were as follows: ferritin: 297 ng/mL, fibrinogen: 356 mg/dL, ALT: 26 U/L, AST: 22 U/L, triglyceride: 164 mg/dL, LDH: 295 U/L, albumin: 23.8 g/dL, D.Bil: 0.3 mg/dL, CRP: 0.2 mg/dL, procalcitonin: 0.14 ng/mL, sodium: 138 mEq/L, and INR: 0.99. The patient was transferred to the Department of Pediatrics. Sequencing of PRF1 from peripheral blood mononuclear cells showed heterozygous mutation (Exon 2, codon 91 GCG/GTG, and Ala/Val).
DISCUSSION
Hemophagocytic lymphohistiocytosis is a clinical entity characterized by increased stimulation and proliferation of benign macrophages responsible for abnormal hemophagocytosis. It has been reported that infections are responsible for 50% of the secondary HLH cases, of which 38% are caused by Mycobacterium tuberculosis and have a higher mortality and a longer prediagnosis symptom duration.
Patients with tuberculosis have increased TNFα, IFNγ, and granulocyte colony-stimulating factor levels compared to healthy population. Specifically, M. tuberculosis could act as an obligate intracellular pathogen after phagocytosis by phagocytic cells to induce TH1-mediated cytotoxicity, activating macrophages and NK cells, further releasing a large quantity of cytokines and chemokines, leading to tuberculosis and HLH-related symptoms.3
Interleukin-10 %3E;60 pg/mL and IFNγ >75 pg/mL have been reported to exhibit 98.9% sensitivity and 93% specificity for the diagnosis of HLH.4 Bone marrow aspiration has a sensitivity of 84% for HLH.5
Padhi et al.,6 in a review involving 63 cases with tuberculosis-induced HLH between 1975 and 2014, reported a mortality rate of 49%. Their patients had a mean ferritin level of 5963 ng/nL, and 54.2% presented with pancytopenia. In univariate analysis, survival was markedly shortened by age more than 30 years, presence of comorbidities, presence of marked hemophagocytosis in bone marrow, and not using or delayed institution of antituberculosis therapy. In our patients, the treatment could not be given regularly because of vomiting in the first week.
Genetic tests in young adults have indicated that mutations or genetic polymorphism may sometimes occur in primary HLH with delayed onset or in secondary HLH.7 Our patient also underwent HLH gene analysis, which was perforin gene mutation.
Hyperferritinemia occurs secondary to ferritin secretion from macrophages and hepatocytes or its release during the erythrophagocytosis process. Lin et al.8 reported that a rapid drop in ferritin level after therapy was associated with a lower mortality. Park et al.,9 on the contrary, reported no association between the rate of ferritin drop and mortality but there was a favorable interaction between a higher fibrinogen level at diagnosis and survival. Our patient had a ferritin level of 11.735 ng/mL and a fibrinogen level of 159 mg/dL, and there occurred a rapid drop in ferritin level.
In tuberculosis-related hollow fiber system, mortality reaches 100% when no antituberculosis treatment is applied while it drops to 40–60% when immunotherapy is combined with antituberculosis therapy. We believe that an early diagnosis in our patient and administration of IVIG, dexamethasone treatments, and simultaneous TPE therapy together with antituberculosis therapy had a role in our patient’s survival.
Therapeutic plasma exchange is an extracorporeal blood purification technique used for the removal of different toxic and inflammatory mediators. Therapeutic plasma exchange can improve clinical and laboratory findings in HLH by reducing circulating inflammatory mediators also including macrophage colony-stimulating factor. Therapeutic plasma exchange also increases response to steroids by reducing IL-6, soluble IL-2, and TNFα levels.10
Hemophagocytic lymphohistiocytosis should be considered in the differential diagnosis of bicytopenia in cases of tuberculosis infection. An early diagnosis and treatment of the condition dramatically reduce its mortality. Therapeutic plasma exchange is a good therapeutic option among cases unresponsive to antituberculosis and immunomodulatory therapy.
AUTHOR CONTRIBUTIONS
Seher Erdoğan and Betül Sözeri developed the concept; Seher Erdoğan and Burcu Karakayalı designed the study; Tuğçe Bozkurt, Betül Sözeri, and Deniz Çakır carried out supervision; Seher Erdoğan and Betül Sözeri provided resources; Burcu Karakayalı and Deniz Çakır provided materials; Seher Erdoğan and Burcu Karakayalı carried out data collection and/or processing; Deniz Çakır, Burcu Karakayalı, and Sevinç Kalın carried out analysis and/or interpretation; Seher Erdoğan and Tuğçe Bozkurt performed literature search; Seher Erdoğan and Sevinç Kalın wrote the manuscript; and Seher Erdoğan and Betül Sözeri performed critical review.
INFORMED CONSENT
Written informed consent was obtained from patient’s parents who participated in this study.
REFERENCES
1. Tseng YT, Sheng WH, Lin BH, Lin CW, Wang JT, Chen YC, et al. Causes, clinical symptoms, and outcomes of infectious disease associated with hemophagocytic lymphohistiocytosis in Taiwanese adults. J Microbiol Immunol Infect 2011;44(3):191–197. DOI: 10.1016/j.jmii.2011.01.027.
2. Bosnak M, Erdoğan S, Aktekin EH, Bay A. Therapeutic plasma exchange in primary hemophagocytic lymphohistiocytosis: reports of two cases and a review of the literature. Transfus Apher Sci 2016;55(3):353–356. DOI: 10.1016/j.transci.2016.09.015.
3. Zhang Y, Liang G, Qin H, Li Y, Zeng X. Tuberculosis-associated hemophagocytic lymphohistiocytosis with initial presentation of fever of unknown origin in a general hospital: an analysis of clinical cases. Medicine (Baltimore) 2017;96(16):e6575. DOI: 10.1097/MD.0000000000006575.
4. Karras A, Hermine O. Hemophagocytic syndrome. Rev Med Interne 2002;23(9):768–778. DOI: 10.1016/S0248-8663(02)00673-2.
5. Ramos-Casals M, Brito-Zeron P, Lopez-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet 2013;383(9927):1503–1516. DOI: 10.1016/S0140-6736(13)61048-X.
6. Padhi S, Ravichandran K, Sahoo J, Varghese RG, Basheer A. Hemophagocytic lymphohistiocytosis: an unusual complication in disseminated mycobacterium tuberculosis. Lung India 2015;32(6):593–601. DOI: 10.4103/0970-2113.168100.
7. Zemed N, Khatib S, Oualil H, Bourkadi J-E. Secondary hemophagocytic lymphohistiocytosis related to hematopoetic tuberculosis. Egypt J Chest Tuberc 2016;65:365–367. DOI: 10.1016/j.ejcdt.2015.12.013.
8. Lin TF, Ferlic-Stark LL, Allen CE, Kozinetz CA, McClain KL. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr Blood Cancer 2011;56(1):154–155. DOI: 10.1002/pbc.22774.
9. Park HS, Kim DY, Lee JH, Lee JH, Kim SD, Park YH, et al. Clinical features of adult patients with secondary hemophagocytic lymphohistiocytosis from causes other than lymphoma: an analysis of treatment outcome and prognostic factors. Ann Hematol 2012;91(6):897–904. DOI: 10.1007/s00277-011-1380-3.
10. Song KS, Sung HJ. Effect of plasma exhange on circulating IL-6 levels in a patients with fatal hemophagocytic syndrome: two case reports. Ther Apher Dial 2006;10(1):87–89. DOI: 10.1111/j.1744-9987.2006.00347.x.
________________________
© The Author(s). 2020 Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and non-commercial reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.