INVITED ARTICLE | https://doi.org/10.5005/jp-journals-10071-23401 |
Fluid Overload and Acute Kidney Injury
1,2Division of Clinical Anaesthesiology, Department of Anaesthesiology, Critical Care and Pain, Tata Memorial Hospital, Homi Bhabha National Institute, Parel, Mumbai, Maharashtra, India
Corresponding Author: Vijaya P Patil, Division of Clinical Anaesthesiology, Department of Anaesthesiology, Critical Care and Pain, Tata Memorial Hospital, Homi Bhabha National Institute, Parel, Mumbai, Maharashtra, India, Phone: +91 9819883535, e-mail: vijayappatil@yahoo.com
How to cite this article Patil VP, Salunke BG. Fluid Overload and Acute Kidney Injury. Indian J Crit Care Med 2020;24(Suppl 3):S94–S97.
Source of support: Nil
Conflict of interest: None
ABSTRACT
Commonest intervention in hospitalized patient is fluid therapy, and practically every critically ill patient receives fluid resuscitation. Commonest indication for fluid administration is to achieve hemodynamic stability and prevent or manage acute kidney injury (AKI). However, fluid administration is a two-edged sword, i.e., inadequate fluids give rise to hypoperfusion and organ injury and overzealous fluid therapy can give rise to fluid overload and related consequences. Though fluids are commonly given to prevent development of AKI, hypervolemia itself has the potential to cause AKI.
Keywords: Acute kidney injury, Fluid movement, Fluid overload, Renal blood flow.
INTRODUCTION
Acute kidney injury (AKI) is commonly encountered in intensive care unit (ICU) patients and is associated with increased morbidity and mortality. Incidence of AKI in critically ill patients is around 5.7% of which almost 50% have sepsis and septic shock.1 Acute kidney injury is a syndrome with multiple etiologies occurring at different clinical settings. Therefore, it is of utmost importance to achieve early diagnosis of AKI; and once diagnosed, it should be promptly evaluated and treated, especially attention should be paid toward reversible causes of AKI. Acute kidney injury can be classified based on the causative factor into intrinsic renal, prerenal and postrenal AKI. Prerenal causes contribute to majority of community-acquired cases of AKI.2
In case of prerenal AKI, fluid resuscitation is the gold standard, but if this resuscitation continues beyond the correction of hypovolemia, then it is associated with increased morbidity, mortality, and length of hospital stay as well as increased risk of AKI.3,4 Several observational studies have demonstrated a correlation between fluid overload and mortality in critically ill patients with acute respiratory distress syndrome, acute lung injury, sepsis, and AKI.5,6 Prowle et al., in a systematic review of 24 perioperative studies on goal-directed fluid therapy (GDT), concluded that GDT was associated with lower risk of postoperative AKI.7 In yet another prospective, observational, multicenter study conducted by Wang et al., of 2,526 patients admitted in 30 ICUs, 1,172 developed AKI during the first 3 days. The mortality was significantly higher in AKI group compared to non-AKI group (25.7% v/s 10.1%, p %3C; 0.001). They also concluded that fluid overload was significantly higher in the AKI group and was an independent risk factor for AKI (odds ratio, OR 4.508, 95% confidence interval, CI 2.9–7.01, p < 0.001), and it also increased the severity of AKI. They also showed that the cumulative fluid balance during the first 3 days was an independent risk factor for 28-day mortality.4 To understand why fluid overload can lead to the development of AKI and other organ dysfunction, we need to understand some basic physiology.
FLUID MOVEMENT ACROSS CAPILLARY ENDOTHELIUM
Movement of fluid between vascular and interstitial compartment is regulated by balance between capillary hydrostatic pressure and plasma oncotic pressure in health. Under normal conditions, balance between these forces results in net flux of fluid to the interstitium known as capillary filtration according to Starling equation which states the following:
where Q is transcapillary filtration volume/second, Pcap is capillary hydrostatic pressure, Pint is interstitial hydrostatic pressure, πp is plasma oncotic pressure, πint is interstitial oncotic pressure, Lp is hydraulic conductivity of membrane, S is the surface area for filtration, and σ is Staverman’s reflection coefficient.
The resultant accumulation of fluid in interstitial space is removed by lymphatics. During inflammation or conditions that alter capillary membrane permeability, there is resultant excess fluid in interstitial space, commonly referred to as interstitial edema. Increased capillary hydrostatic pressure, reduced plasma oncotic pressure, and increased capillary permeability have the potential to increase fluid filtration across capillary membrane and thus lead to edema formation.
ENDOTHELIAL GLYCOCALYX LAYER
The endothelial glycocalyx (EGL) is a carbohydrate-rich layer that coats the endothelial surface of vascular endothelium with average thickness of about 2 μm (0.2–8 μm). This layer plays a major role in net transcapillary filtration and after understanding its role the Starling equation has been modified as:
where πg represents subglycocalyx oncotic pressure.
Many factors damage EGL such as inflammatory cytokines, hyperglycemia, atrial natriuretic peptide (ANP), and ischemia–reperfusion injury. Abnormal blood shear stress as may occur in hypervolemia is also shown to increase glycocalyx shedding.8 Rapid administration of fluid in healthy volunteers resulted in elevated plasma levels of hyaluronic acid, suggestive of damage to EGL and, therefore, can be injurious.9
PROTECTION AGAINST EDEMA FORMATION
While there are factors that increase capillary transmembrane shift of fluid, there are number of safety factors that prevent accumulation of fluid and thus avoid edema formation. Increase in interstitial fluid hydrostatic pressure due to excess fluid filtration, reductions in microvascular surface area for exchange (as may occur with arteriolar vasoconstriction), or increases in lymph flow can limit edema formation. As venous pressure increases, the volume of blood within postcapillary venules, larger venules, and veins increases, and these bulge into the extravascular compartment, thereby raising tissue pressure and thus decreasing fluid shift to interstitium.
In healthy individuals, this margin of safety against edema formation is provided upto venous pressure of around 15 mm Hg, which was first demonstrated in 1932 by Krogh and coworkers.10 Only when venous hypertension exceeds these levels, does gross edema form. However, with other factors such as increased capillary permeability and hypoproteinemia (common in critically ill patients), this margin of safety may be much less.
RATIONALE FOR FLUID THERAPY IN THE SETTING OF ACUTE KIDNEY INJURY
Traditionally, every protocol dealing with hemodynamically unstable patients advocates fluid therapy to restore basic physiological parameters, such as blood pressure, central venous pressure (CVP), and urine output (UO), as these parameters can be readily measured at bedside. Early and adequate fluid resuscitation to stabilize sepsis-induced hypoperfusion is an essential part of hemodynamic resuscitation in the management of these patients. Surviving sepsis campaign recommends initial fluid bolus of 30 mL/kg for these patients.11 With common belief that UO is poor man’s cardiac output monitor, chasing UO is a very frequent phenomenon across multiple clinical settings. In critically ill patients, hypovolemia and hypoperfusion are believed to be major causative factors for AKI. Prolonged renal hypoperfusion also leads to tubular injury, especially the tubule segments of outer medulla, as the renal tubules have limited capacity to use anaerobic metabolism. Aggressive fluid therapy is aimed at reversing renal ischemia, diluting nephrotoxic substances, and preventing tubular obstruction due to casts. Traditionally fluids are the mainstay of treatment, whenever the patient develops oliguria or hypotension. In the presence of absolute or relative hypovolemia, adequate fluid resuscitation is necessary to achieve adequate cardiac output, and renal perfusion in patients with shock.
AKI AND REDUCED RENAL BLOOD FLOW
The reduced renal blood flow (RBF) is considered central to the pathogenesis of septic AKI and fluid therapy. However, contrary to this common belief, experimental data in animal models have shown development of AKI in the presence of renal vasodilation and marked increase in RBF.12,13 Thus, the relationship between volume status and UO is weak, and the administration of intravenous fluid to maintain UO would lead to significant salt and water accumulation in many cases.
PATHOPHYSIOLOGY OF HYPERVOLEMIA
Fluid overload leads to endothelial dysfunction due to inflammation and ischemia–reperfusion injury, causing damage to glycocalyx and capillary leakage. Capillary leakage leads to interstitial edema and at the same time, due to significant loss of volume to interstitial compartment, there is reduction in circulating intravascular volume. This may then lead to reduction in renal perfusion pressure and subsequently to AKI.
Also, interstitial edema leads to impairment in diffusion of oxygen and metabolites from capillaries to tissues. Interstitial edema increases tissue pressure and leads to obstruction of lymphatic drainage and disturbance in cell-to-cell interaction, which will lead to progressive organ failure. Kidney’s ability to accommodate increasing hydrostatic interstitial pressures is limited due to renal capsule, and thus all these effects are more prominently seen in kidney. They cause renal congestion, reduction in renal perfusion and glomerular filtration, and subsequently lead to AKI development.14
Fluid overload is also known to cause distension of atria and stretching of vessel walls, causing release of ANP, which further leads to EGL damage, and cascade leads to AKI (Flowchart 1).
HYPERVOLEMIA, INTRA-ABDOMINAL HYPERTENSION AND ITS EFFECTS on KIDNEY
Massive fluid resuscitation and positive fluid balance are known risk factors for intra-abdominal hypertension (IAH) development.15 Intra-abdominal pressure (IAP) in normal adults ranges from 4 to 7 mm Hg. The IAH is defined as sustained or repeated pathologic elevation of IAP to %3E;12 mm Hg. Sustained elevation of IAP of >20 mm Hg is associated with new organ dysfunction. Studies have shown that IAH is an independent risk factor for AKI development.16,17 Elevated IAP leads to compression of intra-abdominal vessels causing compromised microvascular blood flow and increased renal venous congestion. This results in impaired renal plasma flow and decreased glomerular filtration rate, causing AKI. Association between raised IAP and renal blood flow was shown way back in 1947. In their study on healthy human volunteers, Bradley et al. showed that when IAP was increased to 20 mm Hg, the effective renal plasma flow dropped by 24.4%. Also all the volunteers became oliguric with an average reduction in urine flow of 57.4%.18 As IAP approaches IAH, the renal vein pressures increase, giving rise to intrarenal vascular congestion, which may possibly lead to AKI.19 With further rise in IAP, as it approached nearer the levels of abdominal compartment syndrome, additional factors, such as drop in cardiac output and elevated levels of catecholamine, renin, angiotensin, and other inflammatory cytokines, may further worsen renal function.20–22
Fluid overload can also be the cause of sepsis, which will be an additional contributor toward AKI development. Fluid overload, giving rise to bowel wall edema, leads to impaired gut barrier function and bacterial translocation, which are known to be the major sources of endogenous sepsis. Acute kidney injury is known to be an immunocompromised state and is also associated with cytokine release, insulin resistance, and oxidative stress.23 This further contributes to sepsis.
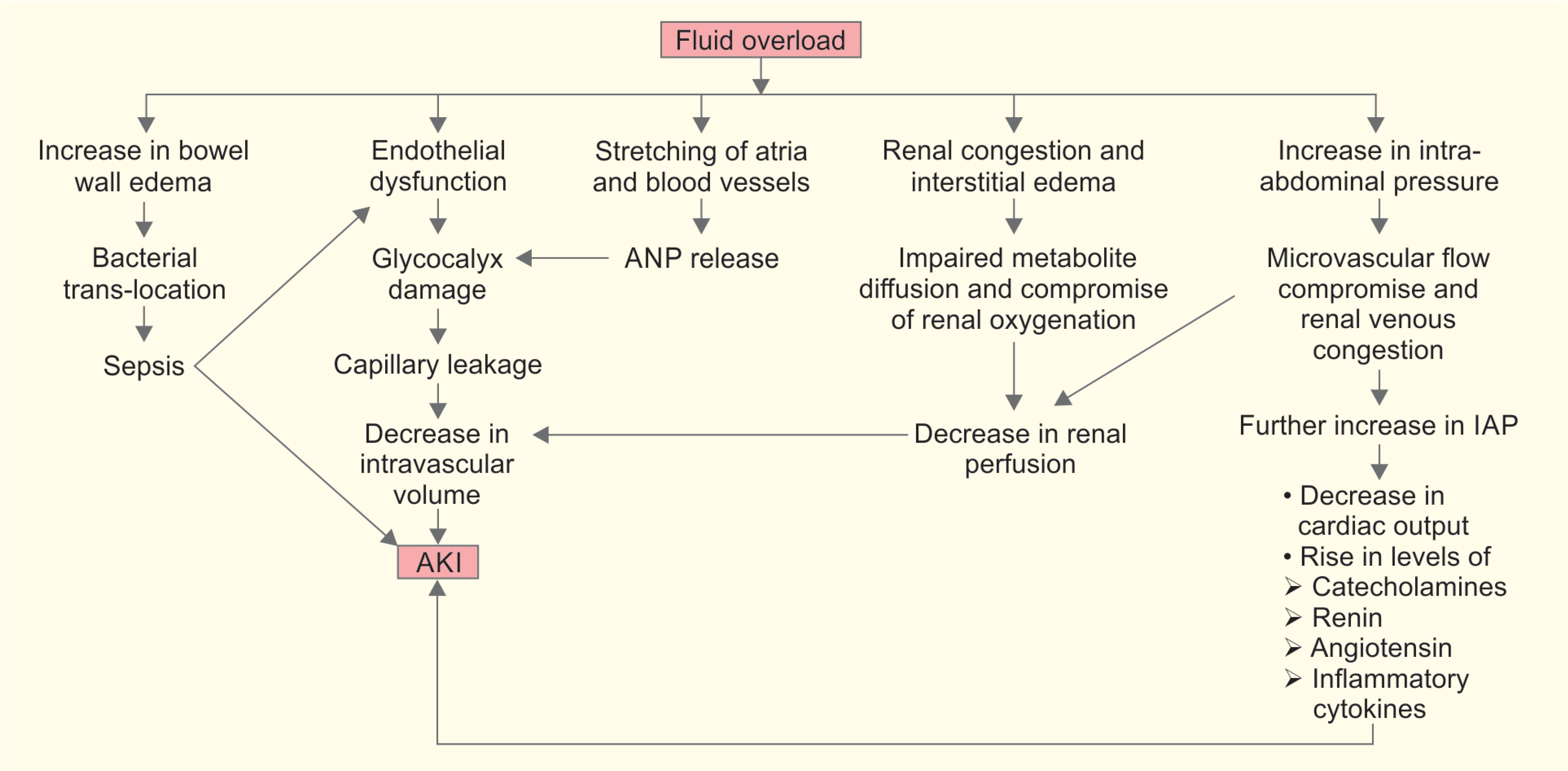
Flowchart 1: Pathophysiology of hypervolemia and AKI
Finally, there is a good physiological rationale and increasing evidence that positive fluid balance is associated with higher incidence of AKI. But it is difficult to say whether AKI is a result of excess fluid therapy or hypervolemia is the result of AKI. In addition, organ cross talk may also play a role in AKI in critical illness; where AKI and edema may be part of critical illness. Patients with severe sepsis tend to have more damage to the EGL, more leaky capillaries, and thus, tend to develop both fluid overload and AKI, following fluid administration.
Fluids should be considered as drugs and should be used judiciously. Accurate assessment of intravascular fluid status, looking for fluid responsiveness, and planning individual targets should be followed in order to improve patient outcomes.
REFERENCES
1. Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, et al. Beginning and ending supportive therapy for the kidney (BEST kidney) investigators: acute renal failure in critically ill patients: a multinational, multicenter study. JAMA 2005;294(7):813–818. DOI: 10.1001/jama.294.7.813.
2. Kaufman J, Dhakal M, Patel B, Hamburger R. Community-acquired acute renal failure. Am J Kidney Dis 1991;17(2):191–198. DOI: 10.1016/s0272-6386(12)81128-0.
3. Ostermann M, Straaten HMO, Forni LG. Fluid overload and acute kidney injury: causes and consequence? Crit Care 2015;19(1):443. DOI: 10.1186/s13054-015-1163-7.
4. Wang N, Jiang L, Zhu B, Wen Y, Xi XM, The Beijing acute kidney injury trial (BAKIT) workgroup. Fluid balance and mortality in critically ill patients with acute kidney injury: a multicenter prospective epidemiological study. Crit Care 2015;19(1):371. DOI: 10.1186/s13054-015-1085-4.
5. Alobaidi R, Morgan C, Basu RK, Stenson E, Featherstone R, Majumdar SR, et al. Association between fluid balance and outcomes in critically ill ChildrenA systematic review and Meta-analysis. JAMA Pediatr 2018;172(3):257–268. DOI: 10.1001/jamapediatrics.2017.4540.
6. Payen D, de Pont AC, Sakr Y, Spies C, Reinhart K, Vincent JL, et al. A positive fluid balance is associated with a worse outcome in patients with acute renal failure. Crit Care 2008;12(3):R74. DOI: 10.1186/cc6916.
7. Prowle JR, Chua HR, Bagshaw SM, Bellomo R. Clinical review: volume of fluid resuscitation and the incidence of acute kidney injury - a systematic review. Crit Care 2012;16(4):230. DOI: 10.1186/cc11345.
8. Chappell D, Bruegger D, Potzel J, Jacob M, Brettner F, Vogeser M, et al. Hypervolemia increases release of atrial natriuretic peptide and shedding of the endothelial glycocalyx. Crit Care 2014;18(5):538. DOI: 10.1186/s13054-014-0538-5.
9. Berg S, Golster M, Lisander B. Albumin extravasation and tissue washout of hyaluronan after plasma volume expansion with crystalloid or hypooncotic colloid solutions. Acta Anaesthesiol Scand 2002;46(2):166–172. DOI: 10.1034/j.1399-6576.2002.460207.x.
10. Krogh A, Landis EM, Turner AH. The movement of fluid through the human capillary wall in relation to venous pressure and to the colloid osmotic pressure of the blood. J Clin Invest 1932;11(1):63–95. DOI: 10.1172/JCI100408.
11. Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Crit Care Med 2017;45(3):486–552. DOI: 10.1097/CCM.0000000000002255.
12. Langenberg C, Wan L, Egi M, May CN, Bellomo R. Renal blood flow in experimental septic acute renal failure. Kidney Int 2006;69(11):1996–2002. DOI: 10.1038/sj.ki.5000440.
13. Langenberg C, Wan L, Egi M, May CN, Bellomo R. Urinary biochemistry in experimental septic acute renal failure. Nephrol Dial Transplant 2006;21(12):3389–3397. DOI: 10.1093/ndt/gfl541.
14. Salahuddin N, Sammani M, Hamdan A, Joseph M, Al-Nemary Y, Alquaiz R, et al. Fluid overload is an independent risk factor for acute kidney injury in critically ill patients: results of a cohort study. BMC Nephrol 2017;18(1):45. DOI: 10.1186/s12882-017-0460-6.
15. Malbrain ML, Cheatham ML, Kirkpatrick A, Sugrue M, Parr M, De Waele J, et al. Results from the international conference of experts on intra-abdominal hypertension and abdominal compartment syndrome. I. definitions. Intensive Care Med 2006;32(11):1722–1732. DOI: 10.1007/s00134-006-0349-5.
16. Sugrue M, Jones F, Deane SA, Bishop G, Bauman A, Hillman K. Intra-abdominal hypertension is an independent cause of postoperative renal impairment. Arch Surg 1999;134(10):1082–1085. DOI: 10.1001/archsurg.134.10.1082.
17. Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P, Acute Dialysis Quality Initiative workgroup. Acute dialysis quality initiative workgroup: acute renal failure - definition, outcome measures, animal models, fluid therapy and information technology needs: the second international consensus conference of the acute dialysis quality initiative (ADQI) group. Crit Care 2004;8(4):R204–R212. DOI: 10.1186/cc2872.
18. Bradley SE, Bradley GP. The effect of increased intra-abdominal pressure on renal function in man. J Clin Invest 1947;26(5):1010–1022. DOI: 10.1172/JCI101867.
19. Doty JM, Saggi BH, Sugerman HJ, Blocher CR, Pin R, Fakhry I, et al. Effect of increased renal venous pressure on renal function. J Trauma 1999;47(6):1000–1003. DOI: 10.1097/00005373-199912000-00002.
20. Winton FR. The influence of venous pressure on the isolated mammalian kidney. J Physiol 1931;72(1):49–61. DOI: 10.1113/jphysiol.1931.sp002761.
21. Firth JD, Raine AE, Ledingham JG. Raised venous pressure: a direct cause of renal sodium retention in oedema? Lancet 1988;1(8593):1033–1035. DOI: 10.1016/s0140-6736(88)91851-x.
22. Butcher BW, Kathleen DL. Fluid overload in AKI: epiphenomenon or putative effect on mortality?. current opinion. Crit Care 2012;18:593–598.
23. Himmelfarb J, McMonagle E, Freedman S, Klenzak J, McMenamin E, Le P, et al. The PICARD group. oxidative stress is increased in critically ill patients with acute renal failure. J Am Soc Nephrol 2004;15(9):2449–2456. DOI: 10.1097/01.ASN.0000138232.68452.3B.
________________________
© The Author(s). 2020 Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and non-commercial reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.